PAIREF - Documentation¶
PAIREF is a tool for macromolecular crystallographers that performs the PAIRed REFinement protocol [1] automatically to estimate the optimal high-resolution cutoff. It is developed in Python 2.7 and can be installed as a module into the Computational Crystallography Toolbox. It provides graphical and command-line interface that executes all the needed calculations. Parameters of refinement can be specified in detail to put all the calculations under full control of the user. Obtained results are presented as plots and tables in HTML log file. PAIREF supports REFMAC5 (part of the CCP4 Software Suite) and phenix.refine (part of the PHENIX) for structure model refinement.
| [1] | P.A. Karplus, K. Diederichs: “Linking crystallographic model and data quality.” (2012) Science, 336(6084):1030-3. |
Examples of resulting graphs:
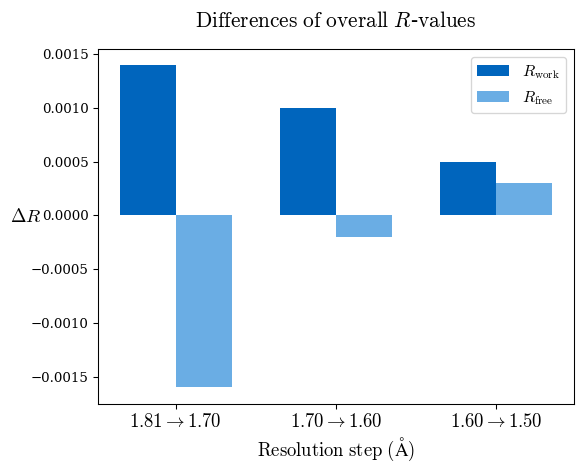
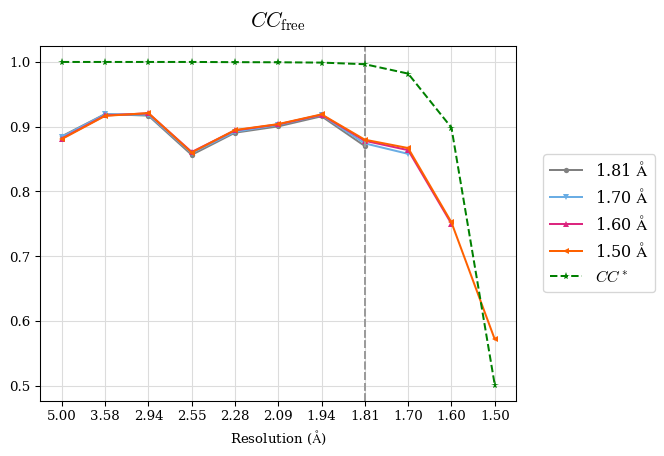
Please reference: M. Maly, K. Diederichs, J. Dohnalek, P. Kolenko: Paired refinement under the control of PAIREF (2020) IUCrJ 7. PAIREF is developed by Martin Malý in collaboration of Czech Technical University, Czech Academy of Sciences, and University of Konstanz. For further information, see Credits.
Getting started¶
PAIREF depends on the CCP4 Software Suite or PHENIX. Both contain the Computational Crystallography Toolbox with Python 2.7.
PAIREF can be easily installed running command cctbx.python -m pip install pairef --user --no-deps in the terminal (GNU/Linux, macOS) or CCP4Console (Windows) or Phenix Command Prompt (Windows). Full instructions are described on the page Installation.
How to use the PAIREF is described on the page Using PAIREF. To run paired refinement of a model (previously refined at 1.81 Å) for a series of cutoffs (1.7, 1.6, and 1.5 Å), execute a following command:
cctbx.python -m pairef --XYZIN model_1-81A.pdb --HKLIN data_1-5A.mtz --HKLIN_UNMERGED data_1-5A_unmerged.mtz -i 1.81 -r 1.7,1.6,1.5