This tutorial shows how to execute paired refinement with PAIREF and interpret gained results. It is assumed that a user has CCP4 suite or PHENIX installed. Follow the instructions in documentation to install PAIREF.
Data: Interferon gamma (POLI)
In this tutorial, we will deal with the data from interferon gamma from Paralichthys olivaceus (POLI). This protein structure was solved at 2.3 Å and is available as PDB entry 6F1E. Detailed information can be found in the related scientific paper 1). Download the archive with the data and extract it in a working folder. The archive contains:
- Structure model previously refined against data at 2.3 Å - poli67_edit12_2-3A_refmac1.pdb.
- Merged diffraction data up to 1.9 Å - poli67_1-9A.mtz
- Unmerged diffraction data up to 1.9 Å - poli67_XDS_ASCII_1-9A.HKL
Note that the structure model poli67_edit12_2-3A_refmac1.pdb was refined in REFMAC5 against work reflections only, i.e. this structure is not the same as the final deposited 6F1E structure that was refined against all the reflections. The X-ray weight was set to 0.06.
Let's have a look on the diffraction data. They exhibited severe anisotropy: According to the criterion of I/sigma(I) > 1.5 in the highest resolution shell, resolution cutoffs could estimated in the range from 2.26 to 2.71 Å. The following table shows merging statistics of poli67_XDS_ASCII_1-9A.HKL calculated with phenix.merging_statistics:
#shell d_max d_min #obs #uniq mult. %comp <I> <I/sI> r_mrg r_meas r_pim cc1/2 cc_ano cc* 01 47.05 5.13 22367 1948 11.48 99.44 494.1 38.8 0.042 0.044 0.013 0.999 -0.214 0.9997 02 5.13 3.63 42232 3343 12.63 99.79 203.3 31.7 0.064 0.067 0.019 0.999 -0.163 0.9997 03 3.63 2.97 57138 4246 13.46 99.95 56.9 14.3 0.151 0.157 0.042 0.997 -0.166 0.9992 04 2.97 2.57 65007 5014 12.97 99.70 11.3 4.0 0.622 0.648 0.178 0.951 -0.025 0.9874 05 2.57 2.30 73887 5623 13.14 99.89 3.7 1.3 1.839 1.914 0.523 0.730 -0.014 0.9187 06 2.30 2.20 37405 2818 13.27 99.93 1.6 0.6 4.462 4.641 1.263 0.400 0.033 0.7559 07 2.20 2.10 45724 3363 13.60 99.91 0.9 0.3 7.623 7.920 2.129 0.196 -0.008 0.5725 08 2.10 2.00 49772 4021 12.38 98.51 0.3 0.1 16.989 17.721 4.963 0.027 0.008 0.2293 09 2.00 1.90 35920 4046 8.88 81.18 0.1 0.0 41.435 43.993 14.417 -0.132 -0.016 undefined
First simple PAIREF run
Now we are prepared to examine data at resolution higher than 2.3 Å. Despite poor merging statistics, there can be still a useful signal in the high-resolution data beyond that cutoff. We will divide the high-resolution data in three shells (2.3-2.10 Å, 2.1-2.0 Å and 2.0-1.9 Å) and add the step by step in the refinement process. Open terminal (GNU/Linux, macOS) or CCP4 console (Windows) and go to the folder where your structure model and diffraction data are saved. For example, if you saved those three files to folder C:/Users/Lab/PAIREF_tutorial_poli, write cd C:/Users/Lab/PAIREF_tutorial_poli into the console and press Enter. Now let's execute PAIREF in a minimalistic way - the required arguments are a structure model and merged diffraction data:
cctbx.python -m pairef --XYZIN poli67_edit12_2-3A_refmac1.pdb --HKLIN poli67_1-9A.mtz -r 2.1,2.0,1.9
Follow the note ------> RESULTS AND THE CURRENT STATUS OF CALCULATIONS ARE LISTED IN A HTML LOG FILE in the program output and open the stated file in your preferred web browser (e.g. Firefox). The running process can be monitored on this HTML page and the results are summarized there as well. You should see a page similar to this. Calculations take some time but they should be finished in 30 minutes.
However, we can see this warning message:
WARNING: The version of REFMAC5 used for refinement of the input structure model `poli67_edit12_2-3A_refmac1.pdb` (5.8.0158) is not the same as the version of REFMAC5 that is now installed and used during paired refinement (5.8.0258).
This indicates that we have to think a bit more about our input structure and data to gain meaningful results. We will have to find out the suitable parameters for this particular case.
Tunning command arguments for particular data
Distant versions of a refinement program such as REFMAC5 can have more or less different behaviour. In this case of structure refined in an older REFMAC version 5.8.0158, it is recommended to refine the structure using the current version at the poorer cutoff (here 2.3 Å) at first. This can be performed with PAIREF: we should add the argument --prerefinement-ncyc 10 so 10 cycles of refinement at 2.3 Å will be carried out before the paired refinement procedure.
Moreover, we must keep in mind the X-ray weight used in previous calculations. In this particular case, we specify it with the argument --weight 0.06 or just -w 0.06.
We can also set a name of our project with the argument --project poli or just -p poli.
CC*, statistic derived from CC1/2, is useful during the results interpretation. For its calculation, the specification of unmerged diffraction data is necessary - then merging statistics will be calculated in the end of the procedure. This can be done with the argument --unmerged poli67_XDS_ASCII_1-9A.HKL or just -u poli67_XDS_ASCII_1-9A.HKL.
So, the final PAIREF command including all the mentioned arguments will be:
cctbx.python -m pairef --XYZIN poli67_edit12_2-3A_refmac1.pdb --HKLIN poli67_1-9A.mtz -u poli67_XDS_ASCII_1-9A.HKL -r 2.1,2.0,1.9 --prerefinement-ncyc 10 -w 0.06 -p poli
In Linux, it is possible to write the same command in a bit clearer way:
cctbx.python -m pairef \ --XYZIN poli67_edit12_2-3A_refmac1.pdb \ --HKLIN poli67_1-9A.mtz -r 2.1,2.0,1.9 \ -u poli67_XDS_ASCII_1-9A.HKL \ --prerefinement-ncyc 10 \ -w 0.06 \ -p poli
If you want to terminate the calculation before it is finished, press Ctrl + C. Within half an hour, the calculations should be done and you will see the results.
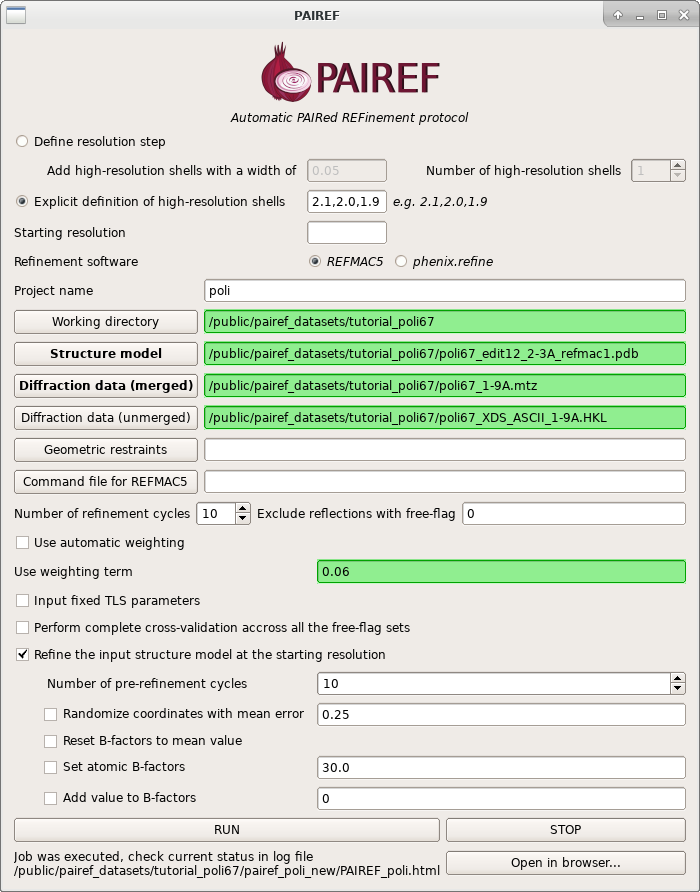
Graphical user interface
PAIREF provides also a graphical user interface (GUI) for intuitive setting of arguments without making up a command. However, it depends on the installation of CCP4 suite. For launching the PAIREF GUI, execute the following command:
ccp4-python -m pairef --gui
To execute the same procedure as in the previous section, set the form analogously to this screenshot and click on the RUN button. After a while, a web page with results is available and you can click on the Open in browser… button.
Interpretation of results - Where to cut?
PAIREF did couple of calculations automatically but the interpretation ofresults is up to us.
The first thing that should be checked is whether the refinements have converged. Scroll at the very bottom of page, here you can see plots of Rwork and Rfree vs. refinement cycle. We can conclude that all the refinements have converged.