PAIREF in CCP4 - CCP4SW Lunchtime Byte
5th Jan 2023
In this short tutorial, we submit a paired refinement job using the PAIREF program, now distributed in the CCP4 suite. You can use our example data from interferon gamma from Paralichthys olivaceus (POLI) 1), PDB entry 6F1E.
Download example data
Download the archive with the POLI data and extract it in a folder - we will refer this folder as working folder. The archive contains:
- Structure model previously refined against data at 2.3 Å - poli67_webinar_2-3A.pdb.
- Merged diffraction data up to 1.9 Å - poli67_1-9A.mtz
- Unmerged diffraction data up to 1.9 Å - poli67_XDS_ASCII_1-9A.HKL
Launch PAIREF from console in working directory
Implementation of PAIREF to CCP4i2 interface is under development but it is not available in the latest CCP4 version 8.0.007 yet. Thus, we will use the standalone graphical user interface (GUI) for PAIREF that can be launched from command line.
In Windows, find the CCP4 console in the Start menu and open it (see the screenshots below). In GNU/Linux or macOS, just open the terminal, assuming all the executables for CCP4 are available there.
Then go to the folder where your structure model and diffraction data are saved using the command cd. For example, if you saved those three files in the folder C:/Users/Lab/PAIREF_tutorial_poli, write cd C:/Users/Lab/PAIREF_tutorial_poli into the console and press Enter.
Now execute the following command for launching the PAIREF GUI:
ccp4-python -m pairef --gui
The structure of POLI was originally refined at 2.3 Å resolution. Nevertheless, we will inspect an impact of the reflection beyond this high-resolution cutoff on the model quality, We have data processed up to 1.9 Å resolution. Thus, we will add three high-resolution shells step by step: 2.3-2.1 Å, 2.1-2.0 Å and 2.0-1.9 Å.
In the PAIREF window, we need to specify:
- How we want to add high-resolution shells - put 2.1,2.0,1.9 to “Explicit definition of high-resolution shells”
- Input structure model
- Input merged diffraction data that contains free flags
- Input unmerged data - recommended, not required
- X-ray weight term 0.06 - from prior knowledge for this particular data set
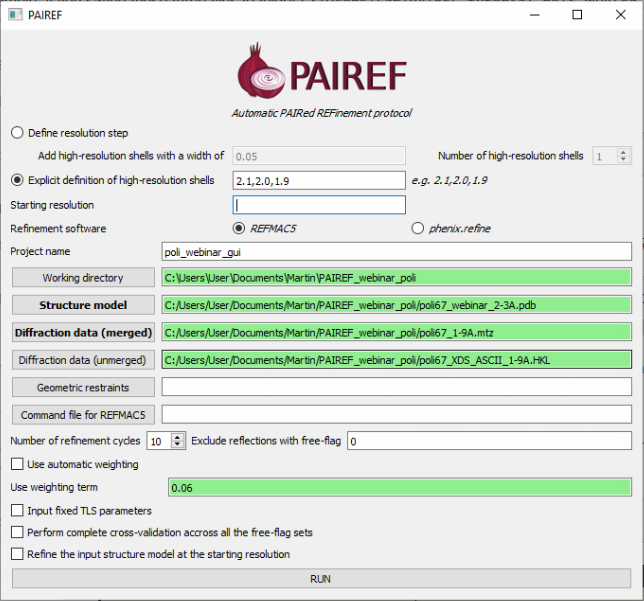
And now we can press RUN and open the HTML log file in a web browser.
Results
The results should look similar to ours: https://pairef.fjfi.cvut.cz/docs/pairef_poli_ccp4sw2023/PAIREF_poli_ccp4sw2023.html.
PAIREF ran all the calculation and did also an automatic suggestion of an optimal high-resolution cut off. Let's check the table on top of the HTML log file:

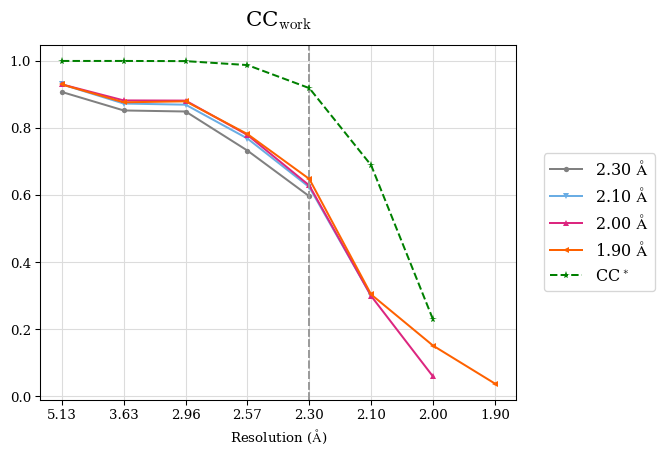
The suggestion is based on the results plotted in the following graphs. Overall Rfree decreased for all the three high-resolution shells that denotes model improvement:
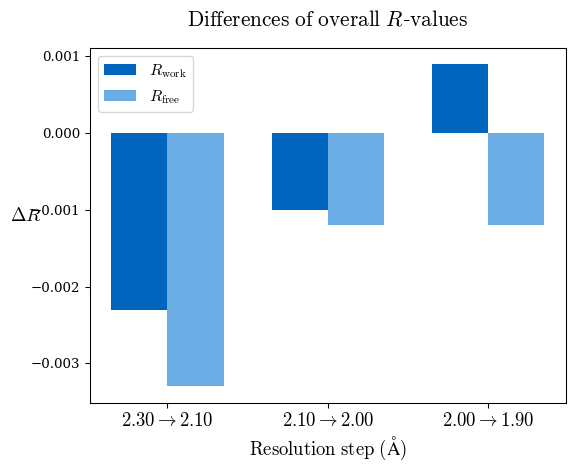
However, we should take into account multiple criteria. Since a perfect model gives an R-value of 0.42 against random data (i.e. pure noise) – assuming non-tNCS (translational non-crystallographic symmetry) data from a non-twinned crystal 2) – a higher R-value in the (current) high-resolution shell indicates either the involvement of high-resolution data without information content (the data are even worse than noise), or poor quality of the model, or the presence of tNCS. This is indicated for the shell 2.0-1.9 Å.
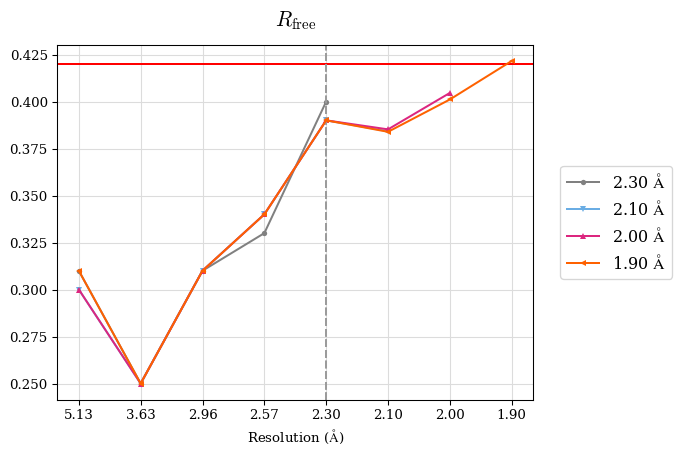
CC* is a model-independent measure of noise is in the diffraction data. For this data set, CC* is higher than CCwork in the whole resolution range, except the shell 2.0-1.9 Å where CC* is undefined due to negative CC1/2. That means overfitting was not indicated but the shell 2.0-1.9 Å should be discarded because these data are very noisy. Note that to access overfitting, it is not needed to use test set, so the comparison of CC* with CCwork is much better than with CCfree as CCwork is calculated on more data.
We should also check whether the refinements have converged. Scroll at the very bottom of page, here you can see plots of Rwork and Rfree vs. refinement cycle. We can conclude that all the refinements have converged, indeed.
We can conclude that the high-resolution limit of the data is ca. 2.1 Å.
Merging statistics:
#shell d_max d_min #obs #uniq mult. %comp <I> <I/sI> r_mrg r_meas r_pim cc1/2 cc_ano cc* 01 47.05 5.13 22367 1948 11.48 99.44 494.1 38.8 0.042 0.044 0.013 0.999 -0.214 0.9997 02 5.13 3.63 42232 3343 12.63 99.79 203.3 31.7 0.064 0.067 0.019 0.999 -0.163 0.9997 03 3.63 2.97 57138 4246 13.46 99.95 56.9 14.3 0.151 0.157 0.042 0.997 -0.166 0.9992 04 2.97 2.57 65007 5014 12.97 99.70 11.3 4.0 0.622 0.648 0.178 0.951 -0.025 0.9874 05 2.57 2.30 73887 5623 13.14 99.89 3.7 1.3 1.839 1.914 0.523 0.730 -0.014 0.9187 06 2.30 2.10 83129 6181 13.45 99.92 1.2 0.4 5.812 6.041 1.633 0.311 0.001 0.6888 07 2.10 2.00 49772 4021 12.38 98.51 0.3 0.1 16.989 17.721 4.963 0.027 0.008 0.2293 08 2.00 1.90 35920 4046 8.88 81.18 0.1 0.0 41.435 43.993 14.417 -0.132 -0.016 N/A
Stay tuned, CCP4i2 interface for PAIREF is coming soon!
Run PAIREF in command line
It is also possible to run PAIREF in the command line. The job described here could be executed using the following command:
ccp4-python -m pairef --XYZIN poli67_webinar_2-3A.pdb --HKLIN poli67_1-9A.mtz -u poli67_XDS_ASCII_1-9A.HKL -r 2.1,2.0,1.9 -w 0.06 -p poli
Contact
In case of any questions or problems, please do not hesitate and write us: m.maly #AT# soton.ac.uk.
Further reading
- More information about PAIREF settings and possibilities are explained in the documentation.
- Linking crystallographic model and data quality. P.A. Karplus & K. Diederichs (2012) Science 336:1030–3
- Assessing and maximizing data quality in macromolecular crystallography. P.A. Karplus & K. Diederichs (2015) Cur. Op. in Str. Biology 34:60–68
- Better models by discarding data? P.A. Karplus & K. Diederichs (2013) Acta Cryst. D59:1215–1222